Montereyのサルマップ2022
 NakagawaShinichi
NakagawaShinichi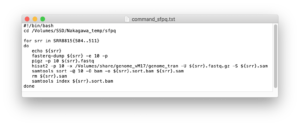
この遺伝子ノックダウンしたらどうなるんだろう?このRNAってこのRNA結合タンパク質についてるんだっけ?というような「わざわざ実験をするのは億劫だけどちょっとだけ知りたい」タイプの疑問に答えてくれる便利なツールがENCODEのページで、最近インターフェースがアップデートされてデータを検索するとマッピングのパターンまで表示してくれるのでだいぶ便利になりました。とはいえ、マウスの細胞株のデータはまだまだ少ないですし、ヒト・マウス以外の動物のデータはカバーされていません。やはりNCBIのSRA searchやDDBJのDDBJ searchページでキーワード打ち込んで、おっ、もうデータあるじゃない、ということで関連sraファイルをまとめてダウンロード、マッピング、IGVでブラウザしてにやにや、、、の「サルでもできるマッピング」の需要は、まだまだあります。最近、立て続けにサルマップする機会があったので、2022年版、アップデートです。
以前サルマップの記事を書いたときはaspera connectとか使っていましたが、ダウンロードのリンク先を見つけるのが一苦労な上、sraファイルからfastqへと変換するところがかったるいというか、やったら時間がかかるのが大きなネックになっていました。ところが今はそんな時代ではなく、fasterq-dumpというツールが超すぐれもの。RunのIDがわかればダウンロードできる上、fastqへの変換も超高速。この手のツールは日進月歩で、たまに知識をアップデートしていかないとあっという間に時代遅れというのを実感。fasterq-dumpについては上坂さんのこちらの記事に詳しいです。
追記:環境によっては、conda listでsra-toolsが入っているのにfasterq-dumpと入れるとfasterq-dump not foundみたいなエラーが出ることがあるようです。その場合はこちらの記事に従ってminiconda/anacondaをアンインストールして、最新版を再インストールすれば良いようです。ただ、この中の「.bash_profile 中の Path を追加する記述を削除する」というところが曲者で、どうやったらいいの?と初歩的なところで困ったときはopen .bash_profileとすればテキストエディットでファイルが開かれるので、適宜miniconda/anacondaに関する記述のところをざっくり削除すればオーケー。
あと、何故か解析に使っていた10年落ちのMacProのご機嫌が相当斜めで、いい子いい子、すねないでね、やればできるからね、となだめすかして使っていたのが、ついにへそを曲げてうんともすんとも言わなくなってしまったので、クリーンインストール、どうせなら最新バージョンでということでMontereyをいれてみました。今のところ特に不具合はないようです。悪名高きsamtoolsの最新バージョンを使うとハマるというdyld: Library not loadedトラップを除いては。。。
まずは環境構築。homebrewでも良いのでしょうが、biocondaのほうが充実著しいので、バッティングを防ぐためconda一本で。まずはLatest versionのminicondaをこちらからインストールしてターミナルでツールを入れていきます。途中、command line tools入ってないけどいれますか、入れなくていいんですか、と聞かれるのでそれはYesと答えておけばオーケーです。
conda config --add channels conda-forge
conda config --add channels defaults
conda config --add channels r
conda config --add channels bioconda
conda install sra-tools #fastx-toolkitが入ってる
conda install hisat2 #マッピング
conda install pigz #gz圧縮用
conda install samtools==1.10 #このバージョンでないとハマる
conda install seqkit #アダプター除去など
あとは、マウスやヒトであればhisat2のインデックスファイルをこちらからダウンロード。インデックスファイルはスプライシングの情報が入ったgenome_tranの方を使わないと、4.5SHがマッピングされないとかいう謎現象が起きたりします。非モデル生物であればインデックスを自前で構築しなくてはいけませんが、スプライス情報を入れたインデックスを作るにはかなりメモリが必要なようです。ここでは、作業ディレクトリを/path/to/working/directoryに、genome_tranから始まるインデックスファイルを/path/to/index/directoryに入れています。******{X..Y}がおとしてきたいSRRファイルのIDを入れるところで、たとえばSRR123456~SRR123462までのRunをマッピングしたければ、SRR1234{56..62}としておけばオーケーです。ドライブのスペースを節約するためにpigzでfastqをfastq.gzに変換しているほか、途中で生成された.samは最後に削除してます。hisat2とsamtoolsをパイプでつなげばいい気もしますが、、、
シングルリードのときは
#!/bin/bash
cd /path/to/working/directory
for srr in SRR*****{X..Y}
do
echo ${srr}
fasterq-dump ${srr} -e 10 -p
pigz -p 10 ${srr}.fastq
hisat2 -p 10 -x /path/to/index/directory/genome_tran -U ${srr}.fastq.gz -S ${srr}.sam
samtools sort -@ 10 -O bam -o ${srr}.sort.bam ${srr}.sam
samtools index ${srr}.sort.bam
rm ${srr}.sam
done
ペアエンドリードのときは
#!/bin/bash
cd /path/to/working/directory
for srr in SRR*****{X..Y}
do
echo ${srr}
fasterq-dump ${srr} -e 10 -p
pigz -p 10 ${srr}_1.fastq
pigz -p 10 ${srr}_2.fastq
hisat2 -p 10 -x /path/to/index/directory/genome_tran -1 ${srr}_1.fastq.gz -2 ${srr}_2.fastq.gz -S ${srr}.sam
samtools sort -@ 10 -O bam -o ${srr}.sort.bam ${srr}.sam
samtools index ${srr}.sort.bam
rm ${srr}.sam
done
で適当な名前でテキストファイルを保存。あとはターミナルでsh 適当な名前.txtで走らせれば自動的にigvで見られるbamファイルが出てきます。マッピング率が低い(90%いかない)ときはアダプター配列が残っていることが多いようです。seqkitなどで除いてやるとだいぶ効率が上がります。最近のdeep sequencingデータはサイズが大きくてfastq-dumpをつかっていたら一晩たっても終わらなかったようなデータも、半日で終わるようになって作業効率が大幅にアップしました。メジャーどころの遺伝子のノックダウン・ノックアウトマウスのデータはかなりの確率でsra searchで見つかるので、ちょっと時間が空いた時にぶらぶら散歩のノリでデータを再解析してみると思いもかけない発見があるやも!?
投稿者プロフィール
最新の投稿
 ノンドメインブログ2025.06.27来し方行末~パート1
ノンドメインブログ2025.06.27来し方行末~パート1 ノンドメインブログ2025.02.11平和な日々
ノンドメインブログ2025.02.11平和な日々 ノンドメインブログ2024.11.25MBSJ2024のシンポジウムのすごいゲスト
ノンドメインブログ2024.11.25MBSJ2024のシンポジウムのすごいゲスト ノンドメインブログ2024.09.17奇遇癖
ノンドメインブログ2024.09.17奇遇癖